Charting changes in a pathogen’s genome yields clues about its past and hints about its future
More than 250 million people worldwide have tested positive for SARS-CoV-2, usually after a diagnostic nose swab. Those swabs aren’t trash once they’ve delivered their positive result, though. For scientists like us they carry additional valuable information about the coronavirus. Leftover material from swabs can help us uncover hidden aspects of the COVID-19 pandemic.
Using what are called phylodynamic methods that can track a pathogen’s travels via changes in its genes, researchers are able to pinpoint factors like where and when outbreaks start, the number of undetected infections and common routes of transmission. Phylodynamics can also aid in understanding and tracking the spread of new pathogen variants, such as the recently detected omicron variant of SARS-CoV-2.
What’s in a swab?
Pathogens, just like people, each have a genome. This is RNA or DNA that contains an organism’s genetic code – its instructions for life and the information necessary for reproduction.
It’s now relatively fast and cheap to sequence a pathogen’s genome. In Switzerland, a consortium of government and academic scientists that we’re a part of as already extracted viral genome sequences from almost 80,000 SARS-CoV-2 positive swab tests.
By lining up genetic sequences obtained from different patients, scientists can see which positions in the sequence differ. These differences represent mutations, small errors incorporated into the genome when the pathogen copies itself. We can use these mutational differences as clues to reconstruct chains of transmission and learn about epidemic dynamics along the way.
Phylodynamics: Piecing together genetic clues
Phylodynamic methods provide a way to describe how mutational differences relate to epidemic dynamics. These approaches allow researchers to get from the raw data about where mutations have occurred in the viral or bacterial genome to understanding all the implications. It might sound complicated, but it’s actually pretty easy to give an intuitive idea of how it works.
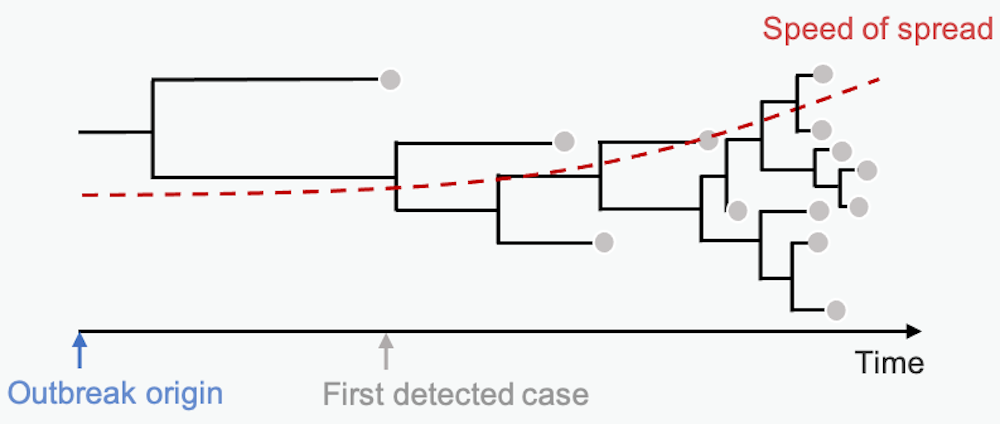
Mutations in the pathogen genome get passed from person to person in a transmission chain. Many pathogens acquire lots of mutations over the course of an epidemic. Scientists can summarize these mutational similarities and differences using what’s essentially a family tree for the pathogen. Biologists call it a phylogenetic tree. Each branching point represents a transmission event, when the pathogen moved from one person to another.
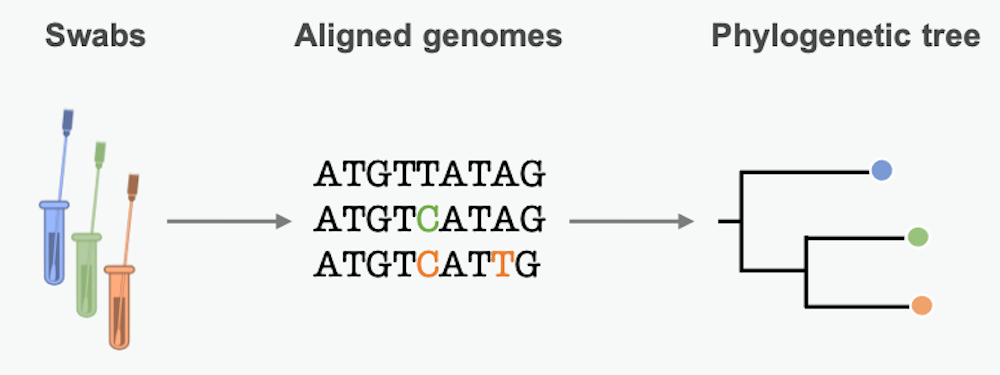
The branch lengths are proportional to the number of differences between sequenced samples. Short branches mean little time between branching points – fast transmission from person to person. Studying the length of branches on this tree can tell us about pathogen spread in the past – maybe even before we knew an epidemic was on the horizon.
Mathematical models of disease dynamics
Models in general are simplifications of reality. They try to describe core real-life processes with mathematical equations. In phylodynamics, these equations describe the relationship between epidemic processes and the phylogenetic tree.
Take, for example, tuberculosis. It’s the deadliest bacterial infection in the world, and it is getting even more threatening because of the widespread evolution of antibiotic resistance. If you catch an antibiotic-resistant version of the tuberculosis bacterium, treatment can take years.
To predict the future burden of resistant tuberculosis, we want to estimate how fast it spreads.
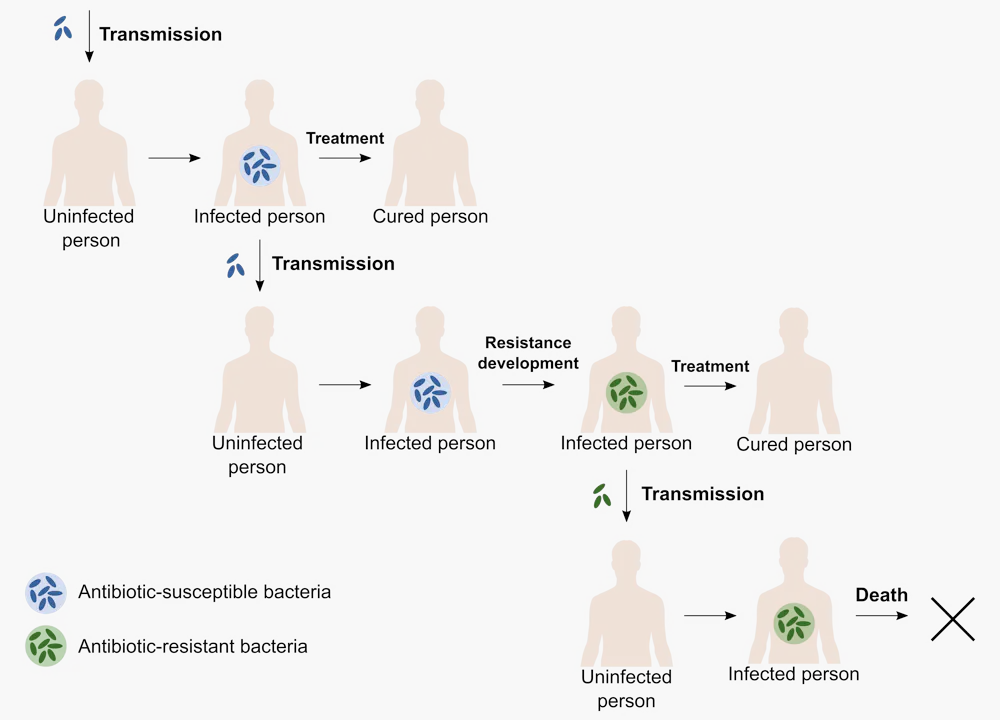
To do this, we need a model that captures two important processes. First, there’s the course of infection, and second, there’s the development of antibiotic resistance. In real life, infected people can infect others, get treatment and, in the end, either be cured or, in the worst case, die from the infection. On top of this, the pathogen can develop resistance.
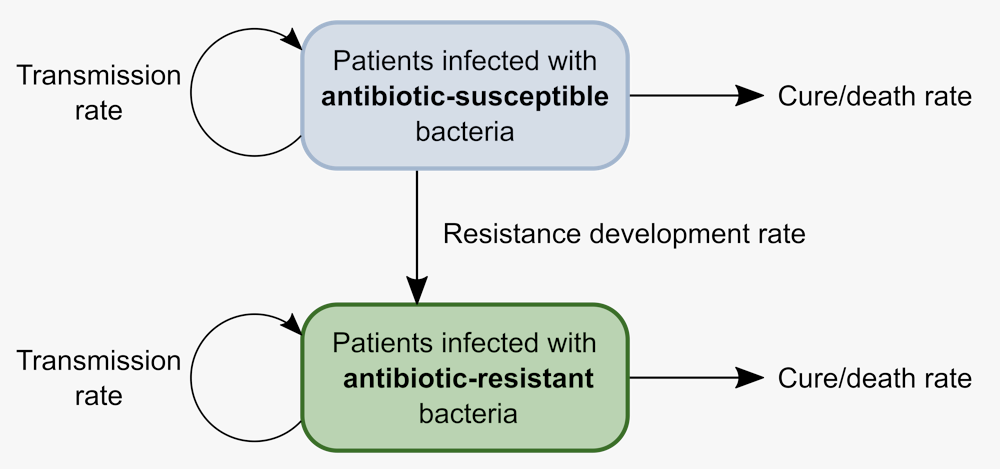
We can translate these epidemiological processes into a mathematical model with two groups of patients – one group infected with normal tuberculosis and one with antibiotic-resistant tuberculosis. The important processes – transmission, recovery and death – can happen at different rates for each group. Finally, patients whose infection develops antibiotic resistance move from the first group to the second.
This model does ignore some aspects of tuberculosis outbreaks, such as asymptomatic infections or relapses after treatment. Even so, when applied to a set of tuberculosis genomes, this model helps us estimate how fast resistant tuberculosis spreads.
Capturing hidden aspects of epidemics
Uniquely, phylodynamic approaches can help researchers answer questions in situations where diagnosed cases do not give the full picture. For example, what about the number of undetected cases or the source of a new epidemic?
A good example of this type of genome-based investigation is our recent work on highly pathogenic avian influenza (HPAI) H5N8 in Europe. This epidemic spread to poultry farms and wild birds across 30 European countries in 2016. In the end, tens of millions of birds were culled, devastating the poultry industry.
But were poultry farms or wild birds the real driver of spread? Obviously we cannot ask the birds themselves. Instead, phylodynamic modeling based on H5N8 genomes sampled from poultry farms and wild birds helped us get an answer. It turns out that in some countries the pathogen mainly spread from farm to farm, while in others it spread from wild birds to farms.
In the case of HPAI H5N8, we helped animal health authorities focus control efforts. In some countries this meant limiting transmission between poultry farms while in others limiting contact between domestic and wild birds.
More recently, phylodynamic analyses helped evaluate the impact of control strategies for SARS-CoV-2, including the first border closures and strict early lockdowns. A big advantage of phylodynamic modeling is that it can account for undetected cases. The models can even describe early stages of the outbreak in the absence of samples from that time period.
Phylodynamic models are under intensive development, continuously expanding the field to new applications and larger datasets. However, there are still challenges in extending genome sequencing efforts to undersampled species and regions and upholding rapid public data sharing. Ultimately, these data and models will help everyone gain new insights on epidemics and how to control them.
ABOUT THE AUTHORS
Claire Guinat, Postdoctoral Fellow in Computational Evolution, Swiss Federal Institute of Technology Zurich. Claire completed her Ph.D. in 2016 in veterinary epidemiology at the The Royal Veterinary College (RVC) and The Pirbright Institute in the UK and then moved to France where she obtained a postdoc fellowship at the Ecole Nationale Vétérinaire de Toulouse (ENVT) and the French National Institute for Agricultural Research and Environment (INRAE). In September 2020, Claire started her Marie Skłodowska-Curie actions Individual Fellowship within the cEvo research group at ETH Zurich (Basel, Switzerland), in partnership with the ENVT-INRAE, joining the DIGDEEP (Digging deeper into genes to track infectious disease outbreaks) project. Her research focus on the development of public and animal health interventions that require detailed knowledge of infectious disease transmission dynamics, in particular how they spread within and between populations. Follow on Twitter @ClaireGuinat
Etthel Windels, Postdoctoral Fellow in Computational Evolution, Swiss Federal Institute of Technology Zurich. Etthel Windels obtained her PhD in 2021 at the University of Leuven (KUL) in Belgium, where she studied the evolution of antibiotic resistance and tolerance through experimental evolution with E. coli. She then moved to Switzerland to work as a postdoctoral researcher within the Computational Evolution group at ETH Zurich. In her current research, she applies phylodynamic models to study antibiotic resistance evolution and spread in TB epidemics.
Sarah Nadeau, PhD Student in Computational Evolution, Swiss Federal Institute of Technology Zurich. Sarah is a researcher in computation evolution, applying statistical models to learn about infectious disease dynamics. Using phylodynamic models, she quantifies epidemic dynamics like the rate of introduction, speed of spread, or the infectious period from pathogen genome sequencing data. So far, she’s worked towards a better understanding of the origins of the European SARS-CoV-2 epidemic, quantifying SARS-CoV-2 transmission in Switzerland, and understanding the viral and human genetic factors that effect the course of HIV infections.
This article is courtesy of The Conversation.